2024年5月13日,深圳大学生命与海洋科学学院、深圳大学血管病中心的苟德明教授团队在Springer Nature出版社旗下期刊《Cellular & Molecular Biology Letters》(中科院1区,影响因子8.3)发表了题为“N6-methyladenosine modification of KLF2 may contribute to endothelial-to-mesenchymal transition in pulmonary hypertension”的研究论文。深圳大学生命与海洋科学学院苟德明教授为通讯作者,医学部康康副教授以及临床医学本科生向晶晶和张兴士为共同第一作者。

肺动脉高压(PH)是一种严重的肺血管疾病,以肺血管重构为特征,最终可导致右心室衰竭和死亡。尽管近年来对PH的认识有所提高,但其具体的发病机制仍不完全清楚。研究表明,肺动脉内皮细胞向间充质细胞转化(EndMT)在PH的血管重构过程中起关键作用。然而,EndMT的调控机制尚不完全清楚。
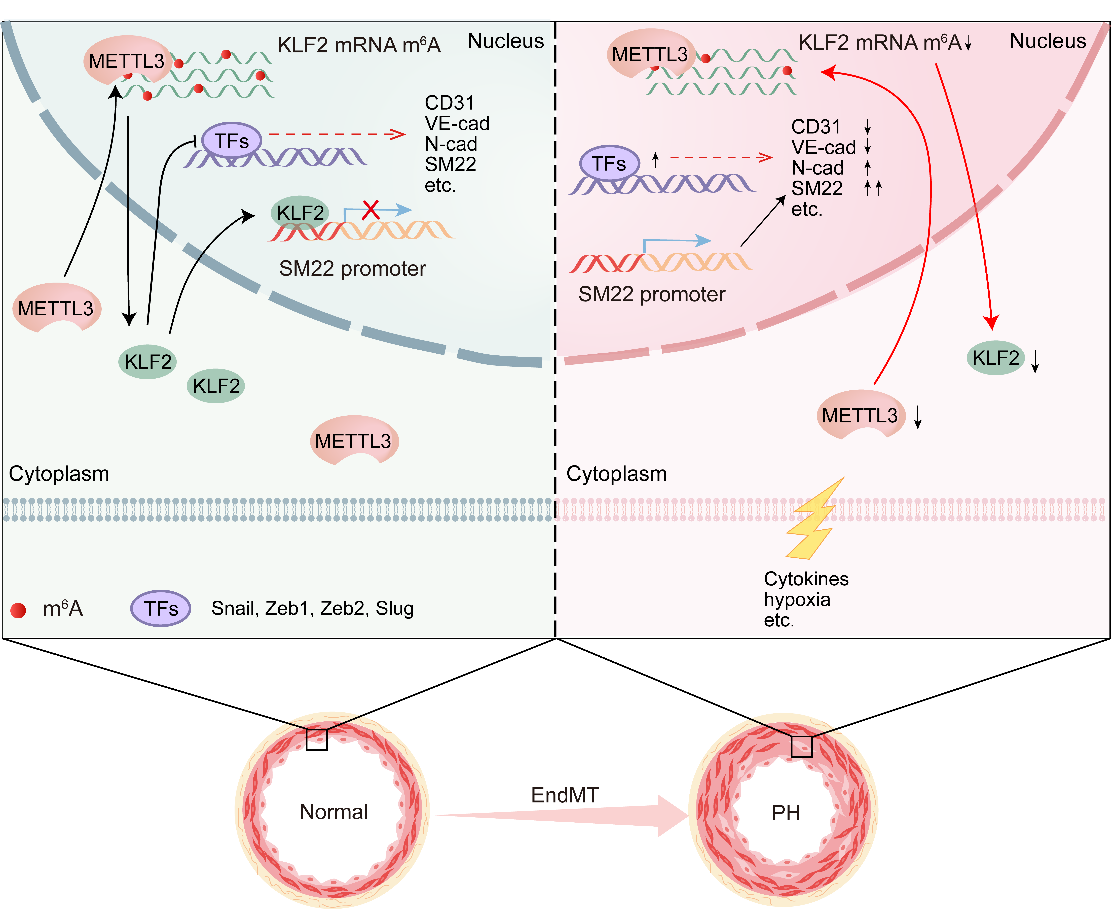
本研究旨在探讨m6A修饰在PH相关EndMT中的作用及其分子机制,特别是METTL3介导的m6A修饰对转录因子KLF2表达的影响。研究团队使用RNA甲基化定量、qRT-PCR和蛋白质印迹等技术,分析了TNF-α和TGF-β1诱导的EndMT过程中m6A修饰和METTL3表达的变化。通过慢病毒介导的基因沉默和过表达技术,评估了METTL3在EndMT中的功能,并利用管腔形成实验和伤口愈合实验评估其对内皮细胞功能的影响。利用内皮细胞特异性基因敲除小鼠,结合血流动力学测量和免疫荧光染色,探讨了METTL3在肺血管重构和PH中的作用。进一步利用RNA-seq、RNA免疫共沉淀qPCR、mRNA稳定性测定和双荧光素酶分析,阐明了RNA甲基化在EndMT中的机制。
研究结果显示,在TNF-α和TGF-β1诱导的EndMT过程中,hPAECs(人肺动脉内皮细胞)的METTL3表达和m6A水平显著下降。抑制METTL3导致内皮标志物(如CD31和VE-cadherin)减少,而间充质标志物(如SM22和N-cadherin)增加,促进了EndMT。在小鼠模型中,内皮细胞特异性敲除Mettl3显著加剧了缺氧诱导的PH症状,包括右心室收缩压(RVSP)和右心肥厚指数(RVHI)的增加,以及肺动脉壁的增厚和重构。进一步研究发现,METTL3通过m6A修饰调控KLF2的表达。KLF2的过表达能够逆转由METTL3抑制引起的EndMT,而KLF2 m6A位点的突变则削弱其对EndMT的保护作用。
这项研究首次揭示了METTL3介导的m6A修饰在PH相关的EndMT中的关键作用,特别是通过调控KLF2表达实现的。这一发现为理解PH的发病机制提供了新的视角,也为未来PH的治疗提供了潜在的新靶点。在临床应用方面,鉴于METTL3/KLF2通路在保护内皮细胞免受EndMT中起关键作用,靶向这一通路可能为PH的治疗提供新的策略,为PH患者带来新的希望。例如,通过开发METTL3的激动剂或KLF2的稳定剂,或直接使用KLF2基因治疗,可能有助于抑制PH的进展。此外,鉴于m6A修饰在多种疾病中的重要性,这一研究也可能对其他涉及内皮细胞功能失调的心血管疾病提供参考。
该研究得到了国家自然科学基金项目(82270054, 82170070, 82241022)、深圳市基础研究面上项目(JCYJ20210324120206017)等课题的支持。
文章链接:https://cmbl.biomedcentral.com/articles/10.1186/s11658-024-00590-w
生命与海洋科学学院
2024年5月16日
 联系我们
联系我们